

-60���늣������ܵ͜�䇽���늳�
�r�g:2020-09-21 11:59��Դ:����ԴLeader ����:�{����
�c��:
��

�҇������|韣��ڱ����^�������ͨ�������͵�0�����£����ֵ^���������͵�-20�����£�������Ҏ����x��늳��wϵ�a���@����Ӱ푡��͜���늽�Һ��늌��ʕ����F�@���Ľ��ͣ�ͬ�rLi+��ʯīؓ�O�еĔUɢ�ٶ�Ҳ����ͣ�����늳��Ĺ������ܮa�������˥����
���գ����������WԺ��Akila C. Thenuwara����һ���ߣ���Matthew T. McDowell��ͨӍ���ߣ��о�����������܄��м����m���ĭh��̼������܄�������FEC��EC�ȣ��܉���Ч������䇽���늳��ڵ͜��µĎ삐Ч�ʺ�ѭ�h���ܡ�
ͨ�����Խ���䇳��e�^�̺�SEIĤ�Ę����ܵ��ضȵ�Ӱ푺ܴ��о������͜��³��e���ɵĽ�����w���ߴ��С���ӄ��˽��渱�������Ķ������ˎ삐Ч�ʵĽ��͡�
��ԓ�о������߰l�Fͨ�^������܄��wϵ������10%���ҭh��̼�����܄�������EC��FEC�ȣ��܉���Ч�������͜��³��e�Ľ�����w����ֱ����ͬ�r����ԓ늽�Һ�wϵ���γɵ�SEIĤ�к����^���LiF��Li2CO3���w�����ڼ�����܄���늽�Һ���γɵ�SEIĤ�tֻ�^�쵽��LiF�ɷ֣��o�C�ɷ��^���SEIĤ�����˽���늳صĵ͜����ܡ�
�Da����DOL/DME=80:20��늽�Һ�wϵ�������˲��ֵ�FEC��EC�����䇌��Q늳���60����-60���µ�ѭ�h���ܣ�늽�Һ�е���}��0.8M LiTFSI����������0.2M LiNO3���ĈD���܉����ڼ�������܄���늽�Һ��60��-0��ķ�����늳صĘO�������^С�����Ǯ��ضȵ���0��r��늉������_ʼ���F���@�IJ��ӣ���������䇳��e�^�̵IJ��������Լ����F������䇡�����늽�Һ������10%���ҵ�FEC��EC����60��-60��ķ����ȶ��]�г��F���@��늉����ӬF����늳��ڵ͜��µĘO�����F�����@�����ӣ��y������늽�Һ�͜��µ�늌����܉�l�F����늽�Һ��늌����Ƿdz��ӽ��ģ�����҂��^�쵽�ĘO�����Ӳ���������늽�Һ�͜�늌��ʵ�Ӱ푡������迹�о������������늽�Һ�м���̼������܄���늳صĽ���늺ɽ��Q�迹�����F���@�����ӡ�
������C̼������܄����Ӱ푽����ؓ�Oѭ�h�^���еĎ삐Ч�ʣ����߲���Li/���P�Ƭ늳��ڲ�ͬ�ض����M����ѭ�h�����Db-d���Կ�����늽�Һ�м���̼������܄�����-20�����r늳صĎ삐Ч������ڼ�������܄������@��������������FEC���ӄ���늽�Һ����-60������Ȼ�܉��_��85%���ϵij�ʼ�삐Ч�ʣ���������܄�늽�Һ�ڴ˜ض��µĎ삐Ч�ʃH��0-20%���ڽӽ��Ҝصėl���£���������FEC��늽�Һ��100��ѭ�h�Ў삐Ч��ƽ����97.6%�������ü�����܄���늽�Һƽ���삐Ч�ʃH��96.3%��������60��ĸߜؗl���£�������܄���늽�Һ���F���˸��õĎ삐Ч�ʣ�50��ѭ�h��ƽ���_��98.7%��������FEC��늽�Һƽ���H��95%�����������܄������Ӳ��ֵ�FEC���܉����䇽���늳صĵ͜�ѭ�h���ܡ�
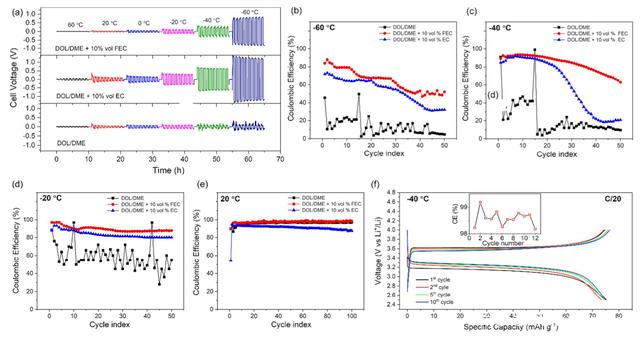
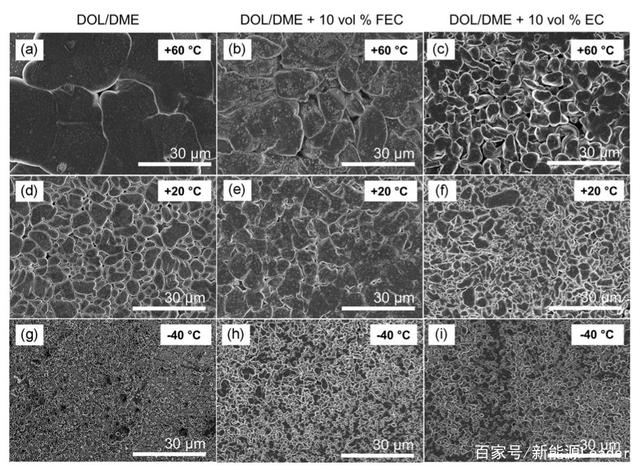
�����Mһ����������FEC������䇽���늳ص͜����ܵęC�������ߌ��ڲ�ͬ늽�Һ��ѭ�h���ؓ�O��ò�M���˷�����ͨ�������҂��J����e�Ľ�����w���w�eԽ�ȱ���eԽС���t�삐Ч��Խ�ߡ����D���܉����ضȌ��ڽ���䇳��e����ò�����@����Ӱ푣��S���h���ضȵĽ��ͣ����e�Ľ�����w��ֱ���@�����͡���60��ĸߜؗl���£�������܄����ؓ�O���e�Ľ�����w�����ƽ����e�_��389um2���ң�������FEC��늽�Һ�г��e�Ľ�����w������e�t��98 um2���ң�����EC�Ąt��78 um2���ң��@�c�҂�ǰ���^�쵽�ļ�����܄���60��ߜؗl���삐Ч�������һ�µġ�
�������^�͵Ĝض��£�20���-40�棩��������FEC늽�Һ�г��e�Ľ�����w���ɞ������20�����_��84 um2���ң���-40�����_��10.0 um2���ң����ڼ�����܄��У�-40���³��e�Ľ�����w����eֻ��3.0 um2���ң�����С�ġ�
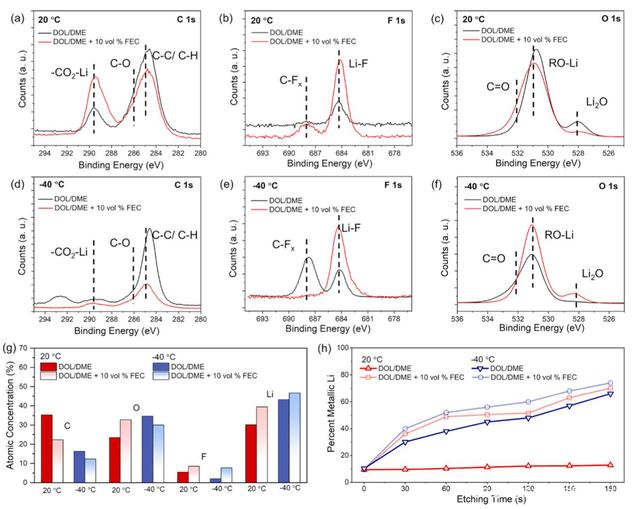
�D�����XPS��������20���-40��l���½���䇱����γɵ�SEIĤ�ijɷַ��������Da-f�܉��ڼ������늽�Һ�У��ڵ͜����γɵ�SEIĤ�к����^�ٵ�̼���}���ЙC�ɷ֡�����FEC��늽�Һ��Ҳ�^�쵽����ƵĬF�������֮�£�����FEC��늽�Һ���γɵ�SEIĤ�к��и����̼���}�ɷ֣�289.7eV�������Db��e��ʾ��F 1s�D�V���܉�������FEC��늽�Һ���γɵ�SEIĤ���и����LiF�ɷ֡���O 1s�D�V���܉����ڵ͜��£�����FEC��늽�Һ���γɵ�SEIĤ�к��и����Li2O�ɷ֡����ā����������늽�Һ������FEC�܄����܉�����SEIĤ�Пo�C�ɷֵĺ��������^�ߵğo�C�ɷ��܉��γɸ������ܵ�SEIĤ�������и��õ��x��늌��ʣ��Ķ���Ч����������䇵Ľ��淀���ԣ�����늳ص�ѭ�h���ܡ�
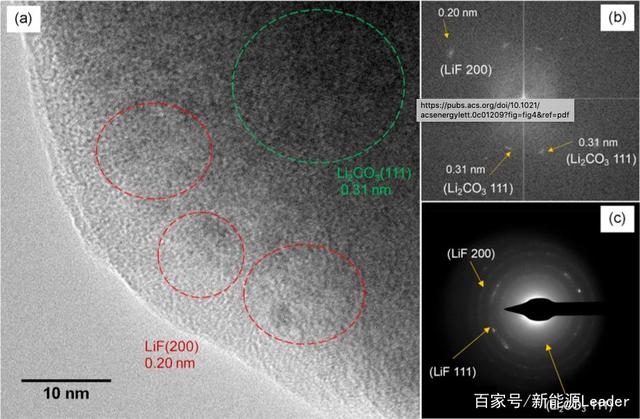
�����Mһ������늽�Һ����SEIĤ�^�Y����Ӱ푣����߲��������R���g��SEIĤ�M���˷������Da��������FEC��늽�Һ�У�-40��l�����γɵ�SEIĤ�Y�����ĈD���܉�SEIĤ�ʬF�Rِ�˵Ę�ʽ��Ҳ���Ǿߺ�䇟o�C���w�w����ɢ�ڟo���ε��ЙC�ɷ��еĽY�������Пo�C�ɷ���Ҫ��LiF��Li2CO3�ɷ֣�����֮ǰ���о��аl�F���ڼ������늽�Һ���γɵ�SEIĤ�еğo�C�ɷ���Ҫ��LiF������LiF���w�w����������С��
�˴Ź����܉��Á�����늽�Һ���܄��Y�����˴Ź����о�����������܄��wϵ��Li+���ӃA���cDME�γ��܄����Y�������Da���Կ���������������܄��м���FEC������Li+�cDME���܄����Y���a���@����Ӱ푣������҂�Ҳע�FECҲ���cLi+�γ��^�����܄����Y����
�����Mһ������FEC����늽�Һ�Y����Ӱ푣����߲��÷��ӄ����W���ߌ�늽�Һ�ڲ�ͬ�ض��µ������M����ģ�M���Dc-fչʾ�˲�ͬ�ض��µ�Li-O�I�돽��Ӌ��Y�������Կ���Li�c��ͬ�܄��γɵ�Li-O�I�ķ�ֵ�����F��2A������Li�cTSFI-��Li-O�I������ߣ��@����TFSI-��Li+�γɵĵ�һ���܄����⚤�а�������Ҫ�����ã����܄�DME��DOL��FECҲ�����c�γ���Li+�ĵ�һ���܄����⚤��Ӌ��ͬ�r�������S��FEC�ļ�����p�ٵ�һ���܄����⚤��DOL�ı������e�ǵ͜����@һ�F��������@���@����FEC���ֵ�������܄����⚤�е�DOL�ɷ֡�
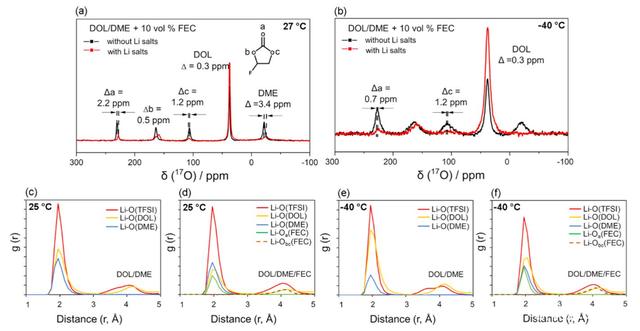
ǰ��ķ�������������FEC��늽�Һ���γɵ�SEIĤ�к���LiF��Li2CO3�ɷ֣������J���@��Ҫ��늽�Һ�нM�ַֽ�늉���ͬ����ģ��о�����LiTFSIֱ�ӷֽ��λ��1.4V���ң��γ�LiF�ķֽ��λλ2.1-2.9V����FECֱ�ӷֽ���λ��0.7V���ң��γ�LiF�ķֽ��λ�t��2.25V���ң�����FEC�cLi+�γ��܄����Y���ֽ��λ�t���Mһ��������0.32V��
Ӌ�������늽�Һ�зֽ�ă������քe��TFSI->FEC>DOL>DME������ڼ�������܄�늽�Һ���γɵ�SEIĤ��ֻ����LiF���@��Ҫ��TFSI-�ֽ�ĽY��������늽�Һ������FEC�t���˕��γ�LiF�⣬FEC�ķֽ�߀���γ�Li2CO3��HCO2Li, Li2C2O4���Լ�LiF�������J���mȻLiF��Li2CO3���x��늌���Ҳ�^�ͣ������@Щ�o�C���w�w���܉���M���g늺ɵĂ��f����Ч�������d���ӵĝ�ȣ��Ķ���Ч�Ĵ��MLi+��SEIĤ���еĔUɢ��
Akila C. Thenuwara���о������ڂ��y������܄�늽�Һ�м��벿�ֵĭh��̼������܄����e��FEC���܉���Ч������䇽���늳��ڵ͜��µĎ삐Ч�ʺ�ѭ�h���ܣ��о������@��Ҫ�����FEC�ļ���ʹ��SEIĤ��������LiF��Li2CO3�ȟo�C�ɷֺ��������M��Li+��SEIĤ���ДUɢ��
������Ҫ���������īI�����H���ڌ����P�ƌW��Ʒ�Ľ�B���uՓ���Լ��n�ý̌W�ͿƌW�о������������̘I��;�������κΰ�����}��Ո�S�r�c�҂�ϵ��
Efficient Low-Temperature Cycling of Lithium Metal Anodes by Tailoring the Solid-Electrolyte Interphase, https://dx.doi.org/10.1021/acsenergylett.0c01209, ACS Energy Lett. 2020, 5, 24112420, Akila C. Thenuwara, Pralav P. Shetty, Neha Kondekar, Stephanie E. Sandoval, Kelsey Cavallaro, Richard May, Chi-Ta Yang, Lauren E. Marbella, Yue Qi, and Matthew T. McDowell
(؟�ξ�������)




��؟�������ăH�������߂����^�c���c�Ї�늳��˟o�P����ԭ�����Լ�����������ֺ̓���δ�����W�C�����������Լ�����ȫ�����߲��փ��ݡ����ֵ��挍�ԡ������ԡ����r�Ա�վ�����κα��C����Z��Ո�x�߃H����������Ո���кˌ����P���ݡ�
�����Wע�� ����Դ��XXX�����Ї�늳��ˣ�������Ʒ�����D�d������ý�w���D�dĿ�����ڂ��f������Ϣ�������������Wٝͬ���^�c�͌����挍��ؓ؟��
������Ʒ���ݡ������������}��Ҫͬ���Wϵ�ģ�Ո��һ�܃��M�У��Ա��҂����r̎����
QQ��503204601
�]�䣺cbcu@m.69gh.com
�����Wע�� ����Դ��XXX�����Ї�늳��ˣ�������Ʒ�����D�d������ý�w���D�dĿ�����ڂ��f������Ϣ�������������Wٝͬ���^�c�͌����挍��ؓ؟��
������Ʒ���ݡ������������}��Ҫͬ���Wϵ�ģ�Ո��һ�܃��M�У��Ա��҂����r̎����
QQ��503204601
�]�䣺cbcu@m.69gh.com
����ϲ�g
-
���݄����аl�o�늳� �������ߡ���늕r�g���̡��ɱ�����
2021-05-18 09:07 -
����ƌW���аl��������ʽ늳ء� ��늕r�g�H10���
2021-05-17 12:21 -
ʥ����cAddionics�����_�l�������̑B늳� �����h���x���늄���܇
2021-04-23 09:50 -
Ampcera�����Ƴ�ȫ�̑B늳ؼ��g �Ɍ��FEV�����ٳ��
2021-04-16 08:40 -
�Ĵ���W����һ����������x��늳��о�����ȡ�����Mչ
2021-04-14 09:13 -
���Ϳ��ГQ늳ؼ������� ����ֱ�����ÿ�������Դ��늄���܇���
2021-04-12 21:48 -
���B������EES�������������߰�ȫ�ԟo����䇜ʹ̑B�ɳ�늳أ�
2021-04-10 15:17 -
���_˹�_�l��һ�N���ڡ�����犃ȡ���ɳ�늵�늳�
2021-04-06 09:45 -
�о��ˆT���ÙC���W���_�l늳ؼ��g ּ���_�l10��犳��늳�
2021-03-15 09:21 -
�о��ˆT�аl�ϳɾۺ���늳����O���� �����ڿ��늳�
2021-01-26 09:04
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|

���}


���P��
-
���݄����аl�o�늳� �������ߡ���늕r�g���̡��ɱ�����
2021-05-18 09:07 -
����ƌW���аl��������ʽ늳ء� ��늕r�g�H10���
2021-05-17 12:21 -
ʥ����cAddionics�����_�l�������̑B늳� �����h���x���늄���܇
2021-04-23 09:50 -
Ampcera�����Ƴ�ȫ�̑B늳ؼ��g �Ɍ��FEV�����ٳ��
2021-04-16 08:40 -
�Ĵ���W����һ����������x��늳��о�����ȡ�����Mչ
2021-04-14 09:13 -
���Ϳ��ГQ늳ؼ������� ����ֱ�����ÿ�������Դ��늄���܇���
2021-04-12 21:48 -
���B������EES�������������߰�ȫ�ԟo����䇜ʹ̑B�ɳ�늳أ�
2021-04-10 15:17 -
���_˹�_�l��һ�N���ڡ�����犃ȡ���ɳ�늵�늳�
2021-04-06 09:45
�����c
-
2020���늳��ИI�о����
2021-05-11 11:24 -
ͻ�l������һ����Iֹͣ���I����ɢ�T����
2021-05-11 10:02 -
4���҇�����늳��b܇��ͬ������134.0%
2021-05-13 08:26 -
��ο���Pack���F䇺���Ԫ����
2021-06-01 09:25 -
�ɳ�������\�՚�늳أ��Mչ�������δ��
2021-05-19 10:59 -
���_�M�h䇘I�ļҵ�
2021-06-03 09:46 -
��늳ػġ����u�����a��늄���܇��܇��Ҫ���������ˣ�
2021-06-01 21:22 -
�P����������x��늳����B�mʽ��ո���ϵ�y���gҎ�����ȃ���ИI�˜ʵĺ���������Ҋ����
2021-05-31 22:53

©2017 ������� 늳��� �A����̩�Ƽ�������������˾ ���k Power by DedeCms
�rֵ�ɾ��ИIƷ�ƣ����\�����ṩ���������YӍ
��ICP��09081210̖
�rֵ�ɾ��ИIƷ�ƣ����\�����ṩ���������YӍ
��ICP��09081210̖
 ��I��̖
��I��̖ �Ź���̖
�Ź���̖